When is a String Like a String?
L. Allison, C. S. Wallace and C. N. Yee. Department of Computer Science, Monash University, Victoria, Australia 3168.
Supported by Australian Research Council grant A48830856
version: 15 Dec 1989
Presented at the International Symposium on Artificial Intelligence and Mathematics (AIM), Ft. Lauderdale, Florida, January 3-5, 1990.
Abstract. The question of whether or not two strings are related and, if so, of how they are related and the problem of finding a good model of string relation are treated as inductive inference problems. A method of inductive inference known as minimum message length encoding is applied to them. It allows the posterior odds-ratio of two theories or hypotheses to be computed. New string comparison algorithms and methods are derived and existing algorithms are placed in a unifying framework. The connection between string comparison algorithms and models of relation is made explicit. The posterior probability of two strings' being related can be calculated, giving a test of significance. The methods are relevant to DNA and to other biological macro-molecules.
Keywords: string, inductive inference, minimum message length, pattern matching, DNA, macro-molecule.
1. Introduction
The inference of similarities, pattern and structure is a hallmark of intelligence. We apply a method of inductive inference known as minimum message length (MML) encoding to problems over strings of characters. The original motivation for this work[1] comes from the analysis of DNA strings. A DNA sequence is a string over the alphabet of four bases {A,C,G,T}. The remarkable success of molecular biologists in sequencing macro-molecules, which is likely to accelerate with the human genome project, has created ample applications. The paper is framed in terms of DNA strings but the techniques apply to strings over other alphabets such as proteins over the alphabet of twenty amino-acids and ascii strings. Strings and languages are central to computer science and related problems occur in file comparison for example.
It is assumed that any one string is random in the sense of Kolmogorov complexity[5,11,14]. This is not quite the case for biological macro-molecules but it is a good approximation to the truth and it is certainly very difficult to encode a typical DNA string in much less than two bits per character. This assumption is not essential to the MML method but makes its application easier.
Even though an individual string is random, it may be clear that two strings are similar in some way. This raises many questions, for example:
Given two strings, with what probability are they related?
If they are related, in what way are they related?
What parameters of the particular model of relation best apply to the two strings?
Given a set of pairs of strings, <Ai,Bi> where Ai is known to be related to Bi, what can be inferred about the model of relation in general?
We give algorithms to solve the first three problems for simple but useful models of relation. We discuss how the fourth problem can be approached. In addition, existing string comparison algorithms are placed in a unifying framework. The relation models considered are relevant to simple models of DNA mutation, but have more general application.
String comparison is used in more complex problems. Notably, several algorithms for the inference of a phylogenetic tree for a set of strings rely on having a matrix of pair-wise distances[3].
To illustrate the MML method in string comparison, consider a transmitter who must send two strings to a receiver. The two parties may previously have agreed on any theories, algorithms and conventions. If the two strings are unrelated, the transmitter can do no better than transmit one string and then the other. If the strings are related there must be some relationship between the characters of the two strings. If the transmitter can find a "good" theory or hypothesis about such a relationship then it may be possible to encode the two strings succinctly. However, for the message to be intelligible it must include details of the theory that have not been agreed on and this tends to increase the total message length. A very detailed theory may allow a very short encoding of the strings proper but itself adds a great deal to the message length. MML encoding defines an optimal degree of detail in this trade-off. The transmitter-receiver paradigm keeps us honest in that anything not agreed to apriori must be included in the message.
If accurate prior probabilities for strings and relations are known, an optimal code can be designed for them. The message length is minus log2 of the probability of the event described. Often such probabilities are not known. However a model having one or more parameters may be agreed on. A message must include the parameter values; the best parameter values give the shortest message. Two hypotheses may be compared on the basis of their message lengths. The more plausible hypothesis leads to the shorter message and the difference in message lengths is minus log2 of their posterior odds-ratio. If a good null-theory can be identified, this leads to hypothesis testing: no hypothesis having a message length longer than the null-theory is acceptable. The natural null-theory here is that two strings are unrelated, requiring about two bits per base for its message.
There are a number of practical and theoretical advantages to MML encoding as a method of inference. It focuses attention on the language being encoded, on what it can express and on the probabilities of messages. If accurate prior probabilities are not known, nevertheless all the techniques and heuristics of data-compression and of coding theory[10,12,25] can be used to get a good code (theory). MML encoding is safe in that the use of suboptimal encoding for a proposed theory cannot make the theory appear more acceptable than it should be. Where parameters over continuous domains are involved, MML encoding defines an optimal degree of precision for their estimates.
Central to the string problems is the definition of relatedness. There can only be one good universal definition and it must be related to the Kolmogorov complexity of a program to produce the two strings. This is not computable in general but fortunately simpler and yet plausible models exist. These are for the most part based on finite-state machines or other simple machines. It turns out that commonly used string comparison algorithms correspond to such models and we have been able to formalise this practice in the MML framework.
The MML method enables two theories or hypotheses to be compared objectively. It does not say how to invent such a theory although algorithms and heuristics may be apparent in some cases. In general, problem knowledge, such as biochemical knowledge, can and should be used to formulate new theories.
Note that the algorithms described here do not actually encode any messages although we often write as though they do. Rather, they calculate only the lengths of messages.
Solomonoff[20] proposed MML encoding as a basis for inductive inference. Wallace and Boulton[22] produced a practical classification algorithm using the technique. A good introduction to the MML technique was given by Georgeff and Wallace[8]. Wallace and Freeman[23] described the statistical foundations of MML encoding. It has had many applications, for example Chong et al[6] have employed it in classifying images of clouds.
In string comparison, Reichert et al[7,15,26] considered the brief encoding of pairs of strings for a single model of relation. Bishop et al[2] gave a maximum-likelihood method to infer the time since two related strings diverged which is very similar to our method for a one-state model. Sankoff and Kruskall[17], Waterman[24] and Bishop and Rawlings[3] give surveys of string comparison methods and applications.
In what follows, many messages must include one or more integers such as string lengths. In order to encode them, a prior probability distribution for integers must be assumed. When there is no information to the contrary, we use Rissanen's[16] universal log2* prior as the default. Some alternatives are discussed in [1].
2. One-State Models
There are at least two general models of relation between strings based on simple translation machines, which may be called the generative and mutation models. In the generative model, a simple machine reads an input string of generation instructions in a special alphabet and outputs the two given strings in parallel. In the mutation model, a different simple machine reads two inputs in parallel. One input is one of the given strings. The other input is a string of mutation instructions in a special alphabet. The machine outputs the second given string. For the restricted relation models we consider, it can be shown that to every generative model there corresponds a mutation model and vice-versa. In what follows we adopt the generative form, but our use of the words `insert', `delete' and `change' for certain generative instructions is common practice inherited from the mutation form. An indel is either an insert or a delete.
generation instructions string A string B ------------ -------- -------- match (A) A A match (C) C C insert(G) G match (T) T T delete(A) A match (C) C C change(G,C) G C match (T) T T An Example.
A sequence of generative instructions represents a specific relation or alignment between two strings.
It is common practice to assign weights or costs to each instruction. The cost of an instruction sequence is the sum of the costs of its instructions. Given strings A and B, an instruction sequence that generates A and B and achieves the minimum possible cost gives an optimal alignment. The minimum cost defines a function D(A,B) on strings. Writing D[i,j] for D(A[1..i],B[1..j]), D can be calculated by the well known dynamic programming algorithm (DPA). Here for simplicity only, we assume that all matches have an equal cost, all changes have an equal cost and all indels have an equal cost.
Boundary Conditions: D[0, 0] = 0 D[i, 0] = indel * i, i=1..|A| D[0, j] = indel * j, j=1..|B| General Step, i=1..|A| and j=1..|B|: D[i,j] = min(D[i-1,j ] + indel, D[i ,j-1] + indel, D[i-1,j-1] + if A[i]=B[j] then match else change) The Dynamic Programming Algorithm.
The choice of minimum made in the general step of the DPA defines a path from D[0,0] to D[|A|,|B|] which gives an optimal alignment.
One choice of costs for the DPA is to cost matches at zero and changes and indels at one. In this case D(A,B) is a metric known as Sellers'[18] metric. If matches cost zero, changes cost one and indels cost a half, then an optimal alignment contains a longest common subsequence (LCS). Common practice in the choice of costs is ad hoc and there is no objective way to judge what are good costs or what particular costs imply about the relation of strings. Here, we relate costs to probabilities of generative instructions. If something is known of these probabilities, this gives an objective way to choose costs. If probabilities are not known, it allows them to be inferred from given strings.
To encode an alignment of A and B, we transmit a string of match, change, insert and delete instructions. Note that the number of instructions must be included in the message. The probabilities of the instructions are required to design an optimal code. Here we assume a simple one-state machine.
p(m) match p(c) change p(i) insert (in A or delete from B) p(d) delete (from A or insert in B) p(m) + p(c) + p(i) + p(d) = 1 symmetry: p(i) = p(d) One-State Machine.
Almost invariably we want p(i)=p(d). The length of (a code word for) an instruction includes a contribution for the characters of the strings. The length of a match or indel is minus log2 of the probability of the instruction plus two bits for the character involved. The length of a change is minus log2 of the probability of a change plus log2(12) bits for the two differing characters. We use these instruction lengths as costs in the DPA.
Given fixed probabilities and two strings, two alignments can be compared on the basis of their message lengths. The more plausible alignment has the shorter message length. The difference in message lengths gives log2 of the posterior odds-ratio of the alignments.
Usually, probabilities of instructions are not known in advance and must then be included in the encoding of an alignment. One method is to use an adaptive code based on running counts. Alternatively, a header to the message states the probabilities used to an appropriate accuracy. The two methods give very similar message lengths[4].
The optimality of an alignment depends on the probabilities used. When these are not known in advance, a search is necessary to find good values. Reasonable initial values are assumed and an alignment, optimal for these probabilities, is obtained. Probabilities for the next step are obtained from instruction frequencies in the alignment. The process converges in a few steps. A small number of initial values must be tried because of local minima.
Note that an arbitrary table of costs for generative instructions implies certain probabilities. These can be discovered by normalisation. The optimal alignments produced by the DPA are invariant if all costs are multiplied by a constant k>0 and if a constant j is added to the indel costs and 2j is added to other costs. Starting from some table of costs, eg. for Sellers' metric, one can choose j and k to give modified costs which give the same optimal alignments and which can be interpreted as negative log2s of probabilities.
normalised Sellers Sellers LCS-metric MML alignment ------- ------- ---------- ------------- match 0 2j 0 -log2(p(m))+2 change 1 k+2j 1 -log2(p(c))+log2(12) insert 1 k+j 1/2 -log2(p(i))+2 delete 1 k+j 1/2 -log2(p(d))+2 Some Specific Costs for the Dynamic Programming Algorithm.
The probabilities must sum to one and give an expression for k in terms of j. There is one degree of freedom remaining which gives a set of probabilities under which the optimal alignments produced are invariant. Exercising this degree of freedom varies the probabilities of alignments but does not change their rank-order. Normalisation enables the probabilities implied by Sellers' metric, for example, to be calculated.
Costs Probabilities j k Cmatch Cchnge Cindel Pmatch Pchnge Pi=Pd 1.2 4.57 2.40 6.97 5.77 0.758 0.096 0.073 1.4 3.48 2.80 6.28 4.88 0.574 0.154 0.136 2.0 1.87 4.00 5.87 3.87 0.250 0.205 0.273 3.0 0.34 6.00 6.34 3.34 0.062 0.148 0.395 Some Instruction Costs and Probabilities Equivalent to Sellers' Metric.
Any similar metric, such as that corresponding to the LCS problem, can be normalised in this way.
For given costs, optimal alignments are not unique in general. All optimal alignments and indeed all non-optimal alignments contribute to the probability of A and B being related in some way. This has been recognised by Bishop et al[2]. Even for two identical strings, eg. ACGT:ACGT, there is a small contribution from sub-optimal alignments and the shorter the strings are the less certain can we be that they are related! In computing an MML alignment, D[i,j] is minus the log2 of the probability that A[1..i] and B[1..j] arose through being related in one most probable way. The `min' in the general step of the DPA corresponds to choosing the maximum probability. If probabilities are instead added, for they correspond to exclusive events, then D[i,j] is the (-log2) probability that A[1..i] and B[1..j], and ultimately A and B, arose through being generatively related in some unspecified way. We call this the r-theory. As before, it is necessary to iterate to discover the probabilities to use in encoding the theory. To do this, frequencies of instructions are computed in D in a weighted fashion according to probabilities of transitions.
The null-theory is that strings A and B arose through being unrelated and this is the complement of the r-theory. The null-theory is encoded by encoding string A and then encoding string B. Note, the lengths of A and B must be included - see later. The odds-ratio of the strings' being related against their not being related is equal to the binary exponential of the null-theory length minus the r-theory length. It is then simple to calculate the posterior probability of the strings being related.
Running the r-theory algorithm in reverse, D'[i+1,j+1] is the (-log2) probability that A[i+1..|A|] and B[j+1..|B|] arose through being related. Thus, D[i,j]+D'[i+1,j+1]-D[|A|,|B|] is the (-log2) probability that the "true" alignment related A[1..i] to B[1..j] and A[i+1..|A|] to B[j+1..|B|], given that A and B are related. A density plot of this function is a good way of visualising possible alignments.
A C G T key MML plus Probability * . * [0..1] bits [1..1/2] A: . * . # (1..2] bits (1/2..1/4] C: . * . + (2..4] bits (1/4..1/16] G: . * . - (4..8] bits (1/16..1/256] T: . * . (8..16] bits (1/256..1/216] r-theory 16.3 bits null-theory 24.6 bits P(related) = 0.99 Alignment Probability Densities ACGT:ACGT
For identical strings, there is near but not absolute certainty of the obvious alignment. For non-identical strings the picture can be more interesting:
A C T A G C T C C A A C C A A * - . . * # + - . . A: - * - . . A: # # # # - . . C: . + * - - . A: + # + + # - . . G: . - * + - - . C: - # + + + # - . . T: . . - * + - . . C: . - # + + + # - . A: . . + * # - . A: . . - # + + + # - C: . - # # # - A: . . - # + + # + G: . - # * - C: . . - # # # # T: . - - * C: . . - + # * ACGTACGT:ACTAGCT AACCAACC:CCAACCAA r-theory 38.9 bits r-theory 44.1 bits null-theory 40.7 bits null-theory 42.9 bits P(related) = 0.78 P(related) = 0.4 Alignment Probability Densities.
Note the area of uncertainty marked by `#' where `CG' is aligned with `GC' for the first pair of strings and the branch for the second pair. The examples above were created to exhibit certain features; in general results for such short strings should be treated very cautiously.
The r-theory message includes the (weighted) number of generative instructions. The null-theory message must include the length of string A and of string B. If these two lengths are encoded independently then it gives a penalty to the null-theory and favours the r-theory for strings of similar length. In some circumstances this may be appropriate but we do not generally consider strings similar only on grounds of length but require similarity of structure also. The generative model with p(i)=p(d) has a built in expectation that the difference in the lengths of A and B is O(sqrt(|A|+|B|)). We build a similar expectation into the null-theory and transmit |A|+|B| using the log2* prior and |A|-|B| using a binomial prior. If p(i)=p(d)=1/2, the r-theory and the null-theory have the same prior over the difference in lengths. When p(i)=p(d)<1/2 the r-theory has a tighter distribution on the difference. Thus extreme similarity of length still favours the r-theory in the absence of genuine structural similarity, but the message length difference caused by this effect is at most O(log(|A|+|B|)) and is only present if abs(|A|-|B|)<<sqrt(|A|+|B|).
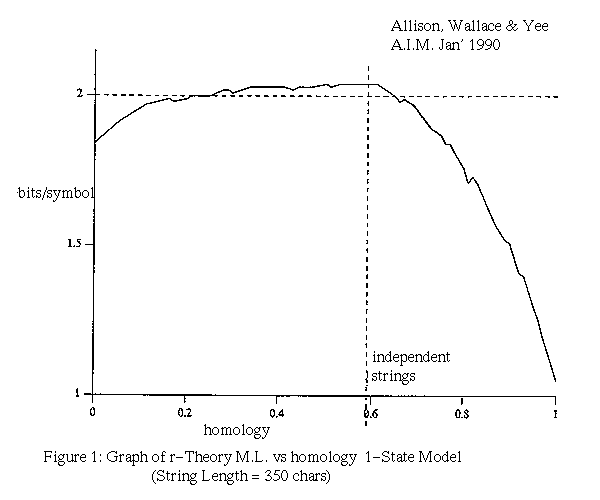
Figure 1 plots the average message length of the r-theory against homology for strings of equal length. The homology is the fraction of characters matching in an optimal alignment (in this case a Sellers alignment) and is commonly used as a measure of similarity. When trying to judge the significance of homology it is common practice to scramble the letters in the two strings and recalculate the homology for a base-line figure. This procedure is not necessary with the MML method which has a built in and rigorous test of significance. The data for figure 1 were generated by mutating strings with homology one (identical) or zero by various amounts using a mutation model in which changes, inserts and deletes occur in the ratio 2:1:1. Note that for long strings, the null-theory is encoded in approximately two bits per character. Note also that very low homology (consider AAAA:CCCC) supports the r-theory because the strings are related in a negative way.
3. Finite State Models
More sophisticated variations on the dynamic programming algorithm are often used in molecular biology. The basic DPA gives a run of L inserts (or deletes) a cost proportional to L. Linear cost functions w(L)=a+b*L are more commonly used for indels. The constants a and b are chosen by experimentation but a=3 and b=1 are typical in work on DNA. As before, matches commonly cost zero and changes cost one. The constant `a' is a start-up cost and discourages large numbers of small indels being inferred. This is found to be plausible biologically. Gotoh[9] gave a fast O(|A|*|B|) algorithm for such costs. In brief, the algorithm maintains three values in each cell of the matrix D. It maintains a value for each possible direction of arrival and a cell can be computed by examining just three neighbours.
The linear costs can be related to probabilities of instructions for a generative machine using the techniques of the previous section. One possible interpretation is a one-state machine with modified insert and delete instructions. These instructions are now taken to insert and delete strings rather than single characters. The alignments produced by Gotoh's algorithm are invariant when the costs are multiplied by k>0 and when 2j is added to every character match and change and jL is added to every indel of length L. Summing the probabilities to one gives an expression for k in terms of j. This enables the probabilities implied by particular costs to be recovered. As before, one degree of freedom remains.
A second interpretation of the linear costs gives a three-state generative machine with simple character instructions. A match or a change always leaves the machine in S1, an insert leaves it in S2 and a delete leaves it in S3. Costs are conditional upon the current state.
S1: match/change-state S2: insert-state and S3: delete-state p(m|Sj) + p(c|Sj) + p(i|Sj) + p(d|Sj) = 1, j=1,2,3 Symmetry: p(i|S1) = p(d|S1), p(m|S2) = p(m|S3), p(c|S2) = p(c|S3), p(d|S2) = p(i|S3) Three-state model.
In general, a three-state model cannot be normalised unless a constant x>0 is subtracted from the cost of entering S2 (and S3) and added to the cost of leaving it. This does not change the alignments inferred. Probabilities of instructions in each state sum to one and this gives two equations in j, k and x. One degree of freedom remains giving a family of values for j, k and x and the implied probabilities.
Costs Probabilities j k x Cmatch Cchnge Cinsrt Cdelet Pmatch Pchnge Pinsrt Pdelet 1.2 3.23 0.29 2.40 5.63 13.84 13.84 0.758 0.242 0.000 0.000 -S1 2.69 5.93 4.43 14.43 0.618 0.197 0.185 0.000 -S2 (~S3) 1.4 2.07 0.63 2.80 4.87 9.05 9.05 0.574 0.411 0.008 0.008 -S1 3.43 5.50 3.47 10.31 0.371 0.265 0.361 0.003 -S2 (~S3) 2.0 0.84 0.79 4.00 4.84 4.59 4.59 0.250 0.418 0.166 0.166 -S1 4.79 5.63 2.84 6.16 0.145 0.242 0.557 0.056 -S2 (~S3) Normalising 3-state model for match=0, change=1, indel(L)=3+1*L
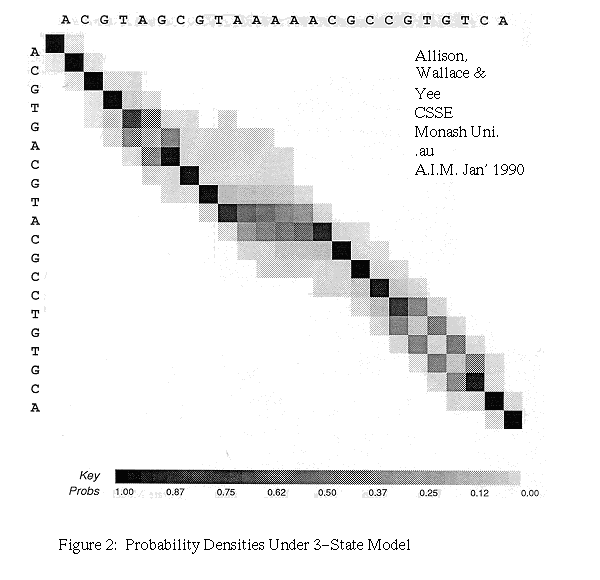
The second interpretation of linear costs as a three-state machine is preferred as it easily allows an r-theory algorithm to be run in a forward and reverse direction and the results to be combined to give a density plot.
In general, the costs in Gotoh's algorithm can be varied to reflect the probabilities of instructions and the algorithm then produces an MML alignment. When probabilities are not known they can be discovered by iteration. We also modify Gotoh's algorithm, rather as the basic DPA was modified, to add probabilities and to produce a weighted sum over all possible alignments. This gives the r-theory of the three-state model. Figure 2 gives an example of a probability density plot.
If any problem-specific knowledge is available, it can and should be used to design new models of relation. First, a language must be designed which has appropriate expressiveness. Second, an optimal (or at least a good non-redundant) coding scheme must be devised for the language. Finally, an algorithm or a heuristic must be designed to infer plausible hypotheses. For example, it is apparent that some pairs of DNA strings have related regions and unrelated regions. The related regions are not necessarily identical. A language to express this possibility might contain character-based instructions as before plus long-insert and long-delete instructions. The latter instructions apply to reasonably large substrings. Assume that long indels are rare events and that their lengths have a geometric distribution with a large mean. The 3-state machine can be augmented with two new states to model this situation.
S1: match/change S2: short-insert state S3: short-delete state S4: long-insert state S5: long-delete state Five-State Model.
The dynamic programming algorithm can be modified to maintain five values - one for each state - in each cell of the matrix D and to sum probabilities much as before. As before, parameter values can be estimated by iteration. The new algorithm now calculates the r-theory for the new model. Note that a run of inserted characters is not classified absolutely as long or short. Rather it is treated in a weighted fashion as both long and short. To be sure, an insert of one or two characters almost certainly implies a short insert and one of many characters almost certainly implies a long insert - almost.
The model just defined also solves the problem of finding substrings or patterns common to two strings. The following special case is of interest in molecular biology and has been examined by Sellers[19] and others:
string A: V;W;X string B: Y;W';Z
W and W' are related but V and Y are unrelated as are X and Z.
4. Other Models
There are a great many models of relation between strings. An important class of model is based on simple finite-state machines. The inference algorithms are variations on the dynamic programming algorithm. Each model is defined by probabilities of machine instructions and by probability distributions on the run-lengths of blocks of matches, changes and indels. The Sellers' and the linear-cost algorithms discussed above imply geometric distributions for run-lengths. A linear cost amounts to using a unary code which is optimal for a geometric distribution. Millers and Myers[13] gave an O(|A|*|B|) alignment algorithm which enables downward-concave cost functions to be used. After normalisation and modulo the characters, a cost function w(L) implies a probability distribution 2-w(L) on lengths. Any problem-specific knowledge of actual probabilities can be built into a cost function. Conversely, if a particular cost function performs well it gives information about mechanisms applying to strings.
Other types of relation model are possible. For example, Tichy[21] gave a linear-time algorithm to find the minimum number of block-moves necessary to edit one string into another. Block mutations such as duplicates, reversals and complements are known to occur in macro-molecules. The MML method allows such rival models to be compared on real strings.
5. Conclusions
We used minimum message length encoding to infer and compare theories over strings. MML encoding makes explicit the connection between a string comparison algorithm and a model of relation. For a given model, the odds-ratio of two alignments can be calculated. The r-theory is the weighted union of all possible alignments. Since there is a natural null-theory, the posterior probability of two strings' being related can be calculated. An algorithm to find substrings or patterns common to two strings was also given. Given enough data, the MML method allows models of relation for "real" strings to be compared and, in principle, the "true" model to be inferred. The methods are clearly applicable to mutating strings such as DNA and other macro-molecules.
6. References
|
These ideas were expanded upon
in later papers, especially: |
[1] L. Allison and C. N. Yee.
Minimum message length encoding and the comparison of macro-molecules.
Bulletin of Mathematical Biology 52(3) 431-453 1990.
[2] M. J. Bishop, A. E. Friday and E. A. Thompson.
Inference of evolutionary relationships.
Nucleic Acid and Protein Sequence Analysis, a Practical Approach
M. J. Bishop and C. J. Rawlings (eds).
IRL Press 359-385 1987.
[3] M. J. Bishop and C. J. Rawlings (eds). Nucleic Acid and Protein Sequence Analysis, a Practical Approach. IRL Press 1987.
[4] D. M. Boulton and C. S. Wallace.
The information content of a multistate distribution.
Journal of Theoretical Biology 23 269-278 1969.
[5] G. J. Chaitin.
On the length of programs for computing finite binary sequences.
Journal of the ACM 13(4) 547-569 1966.
[6] Y. H. Chong, B. Pham, A. J. Maeder and M. J. Marlin.
Automatic nephanalysis using a minimum message length classification scheme.
Australian Joint Artificial Intelligence Conference,
Melbourne Nov 1989.
[7] D. N. Cohen, T. A. Reichert and A. K. C. Wong.
Matching code sequences utilizing context free quality measures.
Mathematical Biosciences 24 25-30 1975.
[8] M. P. Georgeff and C. S. Wallace.
A general selection criterion for inductive inference.
European Conference on Artificial Intelligence 473-482 1984.
[9] O. Gotoh.
An improved algorithm for matching biological sequences.
Journal of Molecular Biology 162 705-708 1982.
[10] R. W. Hamming. Coding and Information Theory. Prentice Hall 1980.
[11] A. N. Kolmogorov.
Three approaches to the quantitative definition of information.
Problems in Information and Transmission 1(1) 1-7 1965.
[12] G. G. Langdon.
An introduction to arithmetic coding.
IBM Journal of Research and Development 28(2) 135-149 1984.
[13] W. Miller and E. W. Myers.
Sequence comparison with concave weighting functions.
Bulletin of Mathematical Biology 50(2) 97-120 1988.
[14] Ming Li and P. M. B. Vitanyi.
Two decades of applied Kolmogorov Complexity.
Proceedings of the Third Annual Conference on Structure in Complexity Theory.
IEEE 80-101 1988.
[15] T. A. Reichert, D. N. Cohen and K. C. Wong.
An application of information theory to genetic mutations and the matching
of polypeptide sequences.
Journal of Theoretical Biology 42 245-261 1973.
[16] J. Rissanen.
A universal prior for integers and estimation by minimum description length.
Annals of Statistics 11(2) 416-431 1983.
[17] D. Sankoff and J. B. Kruskall eds. Time Warps, String Edits and Macro-Molecules. Addison Wesley 1983.
[18] P. H. Sellers.
On the theory and computation of evolutionary distances.
SIAM Journal of Applied Mathematics 26(4) 787-793 1974.
[19] P. H. Sellers.
The theory and computation of evolutionary distances:
pattern recognition.
Journal of Algorithms 1 359-373 1980.
[20] R. Solomonoff.
A formal theory of inductive inference, I and II.
Information and Control 7 1-22 and 224-254 1964.
[21] W. F. Tichy.
The string-to-string correction problem with block moves.
TOPLAS 2(4) 309-321 Nov 1984.
[22] C. S. Wallace and D. M. Boulton.
An information measure for classification.
Computer Journal 11(2) 185-194 1968.
[23] C. S. Wallace and P. R. Freeman.
Estimation and inference by compact coding.
Journal of the Royal Statistical Society series B 49(3) 240-265 1987.
[24] M. S. Waterman.
General methods of sequence comparison.
Bulletin of Mathematical Biology 46(4) 473-500 1984.
[25] I. H. Witten, R. M. Neal and J. G. Cleary.
Arithmetic coding for data compression.
Communications of the ACM 30(6) 520-540 1987.
[26] A. K. C. Wong, T. A. Reichert, D. N. Cohen and B. O. Aygun.
A generalized method for matching informational
macromolecular code sequences.
Computers in Biology and Medicine 4 43-57 1974.
Also see [Bioinformatics] and [MML].
L. Allison, C. S. Wallace and C. N. Yee, 1989; HTML 2001.