Modelling-Alignment for Non-Random Sequences
David R. POWELL, Lloyd ALLISON, & Trevor I. DIX
AI2004, Springer Verlag, LNCS Vol.3339, pp.203-214, 2004.
Abstract. Populations of biased, non-random sequences may cause standard alignment algorithms to yield false-positive matches and false-negative misses. A standard significance test based on the shuffling of sequences is a partial solutions, applicable to populations that can be described by simple models. Masking-out low information content intervals throws information away. We describe a new and general method, modelling alignment: Population models are incorporated into the alignment process, which can (and should) lead to changes in the rank-order of matches between a query sequence and a collection of sequences, compared to results from standard algorithms. The new method is general and places very few conditions on the nature of the models that can be used with it. We apply modelling-alignment to local alignment, global alignment, optimal alignment and the relatedness problem.
Results: As expected, modelling-alignment and the standard PRSS program from the FASTA package have similar accuracy on sequence populations that can be described by simple models, e.g., 0-order Markov models. However, modelling-alignment has higher accuracy on populations that are mixed or that are described by higher-order models: It gives fewer false positives and false negatives as show by ROC curves and other results from tests on real and artificial data.
Availability: An implementation of the software is available here: [www].
Partially funded by Australian Research Council (ARC) grant A49800558.
- Paper:
- [doi:10.1007/978-3-540-30549-1_19]['11].
- Preprint: [PP.ps]
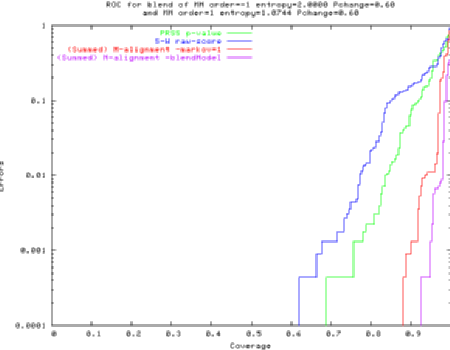
e.g., ROC curve (bottom right is "good"), M-align twice beats PRSS and Smith-Waterman.
Various tests were run against the PRSS (from the FASTA package), Smith - Waterman, and BLAST algorithms, on real and artificial sequences.