The Posterior Probability Distribution of Alignments and its Application to Parameter Estimation of Evolutionary Trees and to Optimisation of Multiple Alignments.
L. Allison & C. S. Wallace,
Department of Computer Science, Monash University, Australia 3168.
Jrnl. Molec. Evol., 39(4), pp.418-430, 1994, doi:10.1007/BF00160274. Also [preprint.ps], and program [source code] with [LogSumData].
This work is partly supported by an Australian Research Council grant.
June 1993, revised March 1994, accepted May 1994.
Abstract: It is shown how to sample alignments from their posterior probability distribution given two strings. This is extended to sampling alignments of more than two strings. The result is firstly applied to the estimation of the edges of a given evolutionary tree over several strings. Secondly, when used in conjunction with simulated annealing, it gives a stochastic search method for an optimal multiple alignment.
Key words and phrases: alignment, estimation, evolutionary tree, Gibbs sampling, inductive inference, Monte-Carlo method, multiple alignment, sampling, stochastic.
Introduction
Estimating the evolutionary "distances" between some given strings, for example DNA sequences, and finding an alignment for them, are examples of inductive inference problems. We rarely know the "right" answer to a question of evolution and must be satisfied with a "good" hypothesis. In previous TARGET="_top"papers (Allison et al 1992a, Yee & Allison 1993), minimum message length encoding was used to infer the relation, if any, between two strings and to compare models of evolution or relation. It is not possible to extend the algorithms given there directly to K>2 strings in any simple way due to rapidly increasing algorithmic complexity. A stochastic process is one possible way around this difficulty. Such a process is described for two problems given a set of K>=2 strings and given an evolutionary-tree over them: firstly estimating the evolutionary "distances" on the edges or arcs of the tree and secondly finding a good alignment of all of the strings. The method relies on sampling from the posterior probability distribution of alignments.
In an inductive inference problem we are given some data D and wish to form a "good" hypothesis H about the data. The language of hypotheses should be tailored to the problem we wish to solve. The following standard definitions are useful:
P(H&D) = P(H).P(D|H) = P(D).P(H|D) -- joint probability
Definitions:
P(H|D) = posterior probability of H given D
P(H) = prior probability of H
P(D|H) = likelihood of H
ML(E) = -log2(P(E)) -- message length of event E
ML(H&D) = ML(H)+ML(D|H) = ML(D)+ML(H|D) -- joint message length
ML(H|D)-ML(H'|D) = ML(H)-ML(H')+ML(D|H)-ML(D|H')
= -log2 posterior odds ratio of H & H'
In the present context, the data consist of some given strings. Alignments and estimates of evolutionary distances are different kinds of hypotheses about those strings. P(D|H) is called the likelihood of the hypothesis H; it is a function of the data given the hypothesis and it is not the probability of the hypothesis. P(H) is the prior probability of the hypothesis H. P(H|D) is the posterior probability of H.
The message length (ML) of an event E is the minimal length, in bits, of a message to transmit E using an optimal code. A long message indicates a low probability and a short message indicates a high probability. It is convenient to work with message lengths, rather than probabilities, for a number of reasons: Typical probability values can be very small and the message lengths are of a more convenient magnitude for handling by computer and by person. The message paradigm reinforces the view that there should be no hidden parameter values associated with the hypothesis itself. All such values are costed explicitly in ML(H). All real-valued parameters must be stated to some optimum but finite accuracy, as described by Wallace and Boulton (1968). In sequence comparison there is a natural null-theory which has a message length being that required to state the given strings individually; this takes approximately two bits per character in the case of DNA. It provides a method of hypothesis testing. The null-theory assumes that there is no pattern or structure in the data. It includes the assumption that an individual string is random. Wallace and Freeman (1987) gave the statistical foundations of minimum message length encoding.
We often wish to find the "best" hypothesis, from a specified class of hypotheses. (Depending on the application, this might be the best evolutionary tree, a good estimate of the evolutionary "distance" between strings, or the best alignment.) It is generally possible to calculate, or at least to give a good approximation to, ML(H) and ML(D|H) under reasonable assumptions. ML(H) can even be ignored if it is a constant for all members of a class of hypotheses. It is not often possible to calculate ML(D) which is unfortunate for it would yield ML(H|D). However by subtracting ML(H'&D) from ML(H&D) it is possible to get a posterior -log2 odds-ratio for two competing hypotheses H and H'; the shorter message indicates the more likely hypothesis. If one hypothesis is the null-theory this also gives the hypothesis test.
An evolutionary tree which is a good hypothesis makes good predictions about the sorts of tuples of characters that occur in good alignments. (A tuple consists of the characters that appear in a column of a conventional alignment.) For example, given four related strings s1 to s4 and assuming that all edges are similar, the tree ((s1 s2)(s3 s4)) predicts that tuples of the form xxyy appear more often than xyxy, whereas ((s1 s3)(s2 s4)) favours xyxy over xxyy. Similarly, a good alignment contains many highly probable tuples which leads to a short message length for the data. In the extreme case of the strings being identical they can all be transmitted for little more than the cost of transmitting one.
An optimal alignment of a set of strings can be used to infer an estimate of evolutionary distances and is sometimes used primarily for that purpose. However a single alignment is a much more detailed sort of hypothesis than an estimate of distances because it also states which characters of the descendant strings are related. In statistical terms, the optimal alignment problem has many nuisance parameters - if the question is one of evolutionary distances. The message for the data given the estimate of distances should not be based on one alignment. It is not the matter that distance estimates are better than alignments or vice-versa; they are answers to different questions. Alignments are useful in their own right, for some purposes.
Yee and Allison(1993) showed that in order to obtain an unbiased estimate of the evolutionary "distance" between two strings it is necessary to use a weighted average of estimates from all alignments whereas the use of a single optimal alignment gives a biased estimate. The average can be computed in a few steps, each step taking time proportional to the product of the string lengths. This is feasible for two strings but not more unless the strings are short. In this paper, a stochastic process is used to average over not all but many alignments of K strings so as to get the estimates of the distances on the edges of a tree over the strings in an acceptable time. This is an example of Gibbs sampling or a Monte-Carlo method (Hastings 1970). Random alignments are generated from the posterior probability distribution of alignments. When used in conjunction with simulated annealing this also gives a stochastic search process for a good alignment. We use tree-costs because these correspond to explicit evolutionary hypotheses; the edges of a tree are modelled individually. We note that Lawrence et al (1993) describe a Gibbs sampling strategy for finding ungapped signals in a set of protein sequences. That work relates each protein to a central model which implicitly represents the constraints on the typical member of the set. It is using a form of star-costs which is probably more suitable for proteins, particularly if they are only distantly related.
There is important prior work in the treatment of alignment and evolutionary trees as inductive inference problems. Bishop and Thompson (1986) first cast pairwise-alignment as a maximum-likelihood problem, summing the probabilities of all alignments. Thorne et al(1991, 1992) extended the maximum likelihood method to more general models of evolution, including conserved and variable regions, and related probabilities of mutation to time. Allison et al (1992a) included the model or hypothesis cost in pairwise-alignment and compared evolutionary models of different complexities on an equal footing. Felsenstein (1981, 1983) treated the inference of an evolutionary tree from a given multiple alignment as a maximum likelihood problem. All these are part of a large and important trend to make models of evolution explicit for better scrutiny and to place subsequent inferences on a sound statistical footing.
Models of Evolution
We model the evolution of a parent string A into a child string B by a finite-state mutation-machine. Such a machine can copy a character, change a character, insert a character or delete a character. Inserts and deletes are collectively called indels. The machine reads A and a sequence of mutation instructions and produces B.
If we wish to consider A and B to be of equal standing, we model their relation under a finite-state generation-machine. Such a machine can generate the same character in A and in B (match), generate different characters in A and in B (mismatch), generate a character in A only (insA) or generate a character in B only (insB). The machine reads a sequence of generation instructions and produces A and B.
Traditionally, a (pairwise) alignment is used to show a hypothetical relationship between two strings. Each string is padded out with zero or more null characters `-' until they have the same lengths. The padded strings are then written out one above the other. The two characters in a column are called a pair, a two-tuple, or just a tuple. The all-null tuple is not allowed. It is convenient to write a pair as <x,y> or just `xy' in text.
There is an obvious correspondence between alignments of two strings, sequences of generation instructions and sequences of mutation instructions, as illustrated in figure 1, and they will be used interchangeably as convenient. Given a finite-state generation-machine, say, the probability of a particular sequence of instructions of a given length is the product of the instructions' individual probabilities and the message length of the sequence is the sum of their individual message lengths. This depends on P(match), P(mismatch), P(insA) and P(insB) which are called the parameters of the machine. The parameters might be given apriori, but in general they must be estimated from the strings. They correspond to the evolutionary "distance" of the strings. Note that an estimate can be got by counting instructions in one, or more, instruction sequences. The parameters consist of several values; a machine has more than one degree of freedom. If it is necessary to reduce them to a single number representing, say, the number of years since divergence, then a rates-model in the style of Thorne et al(1991, 1992) could be fitted to them in some way. Our use of machines differs from the use of hidden Markov models by Haussler et al (1993) in that a machine models an evolutionary process rather than a family of sequences or the typical member of a family. We believe that the former use best captures evolutionary relations and the latter best captures functional constraints.
The advantage of using finite-state machines is that there are standard techniques for manipulating them. The composition of two or more such machines is yet another one. Figure 2 illustrates the evolution of string A from string P as modelled by a mutation machine mA and of B from P as modelled by a mutation machine mB. If we know the parameters of mA and mB then we can calculate the parameters of an equivalent generation machine m=mA.mB which relates A and B directly. There are a number of possible explanations for each generation instruction of m in terms of instructions for mA and mB. We expect mA and mB to execute more instructions than there are characters in P because each copy, change and delete acts on one character of P but an insert does not. The dashed quantities, pA'(c) etc., are the rates of instructions per character of P. Summing the rates of all explanations for all of m's instructions, we get a rate of (1+pA'(i)+pB'(i)) instructions per character of P for m, the excess over 1 being due to the insertion operations of mA and mB. A further normalisation step gives the parameters of m. Similar calculations are used when several machines are combined in the alignment of more than two strings in section 4. Note that we do not allow `x<---_--->y' to be an explanation of xy, for example, because the x and the y are not related in the explanation.
The dynamic programming algorithm (DPA) can be used to compare two strings under a variety of costs calculating, for example, the edit-distance as in figure 3a. We take an optimal alignment to correspond to a most probable instruction sequence. An instruction sequence having the highest probability, and the minimum message length, can be found by a DPA using costs based on the message lengths of instructions, as in figure 3b. (Strictly, a term should be included for the number of instructions in the sequence but all plausible sequences have similar numbers of instructions and the -log2 of those numbers are very similar.) The alignment can be recovered by a traceback through the D[,] matrix or by Hirschberg's (1975) technique. If on the other hand the object is to infer the machine parameters, rather than an optimal alignment, something slightly different should be done. There are many optimal, near-optimal and not so optimal instruction sequences or alignments in general. An estimate, E, of the machine parameters is itself a hypothesis. The best estimate minimises ML(E)+ML(A&B|E), the parameters being stated to some optimal and finite accuracy. ML(A&B|E) is not the message length of any one instruction sequence. It is the -log2 probability that the machine generated strings A and B, ie. the -log2 of the sum of the probabilities over all possible instruction sequences or alignments that yield A and B. The hypothesis is that A and B are related by the inferred machine in some unspecified way and it is called the r-theory, the `r' standing for related. ML(A&B|E) can be calculated by a modified DPA which uses `logplus' instead of `min' in its general step, as in figure 3c, effectively adding the probabilities of alignments. For example, logplus(m,m) = m-1. We now have D[i,j] = ML(A[1..i]&B[1..j]|E). Note that two different alignments represent exclusive explanations and their probabilities can therefore be added. Full details are given in Allison et al (1992a). A code for efficiently transmitting A and B given E, using ML(A&B|E) bits and not based on a single alignment, can be devised but it is sufficient just to know what the message length would be in such a code.
The machine parameters must be estimated if they are not known apriori. An iterative approach is used; it may give only a local optimum. Assume we require an optimal alignment. Initial parameter estimates are "guessed". An alignment is found that is optimal for the current estimates using the DPA of figure 3b. New estimates are derived from the alignment. The process is iterated until it converges, which it must, invariably in a few steps. If on the other hand the objective is to estimate the machine parameters, rather than to find an optimal alignment, a similar approach is used but with the DPA of figure 3c and this is further modified to accumulate weighted averages of instruction frequencies when alignments are combined in the general step. These values are used in the next iteration.
In what follows we consider only the simplest model of evolution, the 1-state model, where the probabilities of instructions are independent of context. In particular, linear indel or gap costs require at least a three-state model. It is also assumed that P(insert) = P(delete) and that all characters are equally probable, all inserts are equally probable and all changes are equally probable. In that case, we have for DNA and the generation machine:
ML(match(x)) = -log2(P(match))+log2(4) = -log2(P(match))+2 ML(mismatch(x,y)) = -log2(P(mismatch))+log2(12) ML(insA(x)) = ML(insBgp(x)) = -log2(P(insA))+2 = -log2(P(insB))+2These assumptions are made in the interests of simplicity as we are primarily interested in the underlying algorithms and methods. The model is simple but is not unacceptable for DNA. A more general model of changes would have to be incorporated if the methods were to be used on protein sequences. It is in any case the indels that are the main source of algorithmic complexity in string comparison.
The Posterior Distribution of Alignments
The stochastic processes to be described are based on a method of sampling alignments (or machine instruction sequences), given two strings A and B, with the correct frequency as determined by the posterior probability distribution of alignments. A sampling procedure that achieves this aim is sketched below with more details being given in the appendix. It is extended to multiple alignments of more than two strings in later sections. It strongly resembles Hirschberg's (1975) linear-space version of the DPA in its use of a recursive divide-and-conquer technique.
Assume that A is of length m and B is of length n. Informally, the procedure divides A at i=m div 2 into A1=A[1..i] and A2=A[i+1..m]. It then chooses a point, j, and divides B into B1=B[1..j] and B2=B[j+1..n]; j might or might not equal n div 2. The procedure is then called recursively on A[1..i] and B[1..j] and on A[i+1..m] and B[j+1..n] until the base case is reached. The base case is that A contains just one character. It is straightforward to enumerate all the allowable ways of generating A and B in the base case and to sample from them according to probability. The choice of j in the general case is made according to the probability distribution of values taken over all alignments or instruction sequences. The DPA of figure 3c calculates the probability of A and B being generated - by summing over all possible alignments of A and B. (It actually works with the -log2 of probabilities.) It can also be used to calculate the probabilities of A[1..i] and each prefix of B being generated; these values are contained in one row of D[,]. Running the DPA "in reverse" gives the probabilities of A[i+1..m] and each suffix of B being generated. These results are combined and normalised to give the probability distribution of j given i, and j is sampled accordingly. This is closely related to the notion of alignment density plots in Allison et al (1992a, fig 6, fig 11).
The complete sampling procedure is just twice as slow as the DPA that it uses, ie. it takes O(|A|×|B|) time. This is because it solves one full-size (|A|×|B|) DPA problem, two problems whose total size (|A|×|B|/2) is half that of the original problem, four problems whose total size is one quarter that of the original problem etc. In Hirschberg's algorithm the divide-and-conquer technique was used to reduce the use of space to O(|B|) - as it still does here - because only two rows of a matrix of length |B| are required to calculate the values needed.
K >= 2 Strings
A (multiple) alignment of K strings is formed by padding out each string with zero or more null characters so that they all have the same lengths. The padded strings are then written out one above another. Each column of characters is called a K-tuple or just a tuple. The all-null tuple is not allowed. There are two parts to the optimal alignment of several strings. The first is the search algorithm for finding a good alignment. The second is the cost function to be applied to alignments. As before, we take an optimal alignment to be a most probable alignment of the strings.
The naive extension of the DPA to K strings, each of approximately n characters, would require O(2KnK) running time which is infeasible for even modest values of K and n. However the DPA can be extended so as to align two alignments. A string can be considered to be a trivial case of an alignment and its characters to be one-tuples. Given an alignment AS over a set of L strings S and an alignment AT over a set of M different strings T, AS and AT can be aligned to give an alignment of K=L+M strings S ∪ T. The algorithm aligns AS, a string of L-tuples, with AT, a string of M-tuples. All that is necessary is that a cost be given to each K-tuple within the DPA; this is described below. The final alignment may not be optimal for the L+M strings but this algorithm can be used as an iterative step to improve a multiple alignment to at least a local optimum. This kind of deterministic heuristic is quite common and an example has been described by Allison et al(1992b): Given K>2 strings and an evolutionary tree over them an initial K-way alignment is found by some suboptimal process. The tree is then "broken" on some edge which partitions the strings into two disjoint sets, S of size L and T of size M=K-L. The K-way alignment is projected onto these two sets of strings to give two sub-alignments, AS over S and AT over T, which are re-aligned with the DPA to give a new overall K-way alignment. The process is iterated and terminates when there is no further improvement in the full K-way alignment. The results are usually good although it is not guaranteed to give an optimal K-way alignment. It may get stuck in local optima and results may depend on the initial alignment and the order in which edges are chosen during improvement. The alignment sampling process of the following section provides a way around these and other difficulties.
The other part of the optimal alignment problem is the assignment of a cost to an alignment of K strings. We use tree costs and model each edge of the evolutionary tree by a separate mutation-machine. The machines can be combined so as to calculate the probability, and hence the message length, of each K-tuple examined by the DPA. The DPA can then find a good K-way alignment and calculate its total message length by summing K-tuple message lengths. A particular K-tuple of descendant characters can have several evolutionary explanations. If we knew the hypothetical ancestral characters at the internal nodes of the tree it would be a simple matter to combine the probabilities of the implied machine instructions on the edges. Since we do not, it is necessary to sum over all possible assignments to internal nodes in the style of figure 2. Fortunately the combinatorial possibilities can be dealt with efficiently; Felsenstein(1981, 1983) describes the necessary algorithm. It involves calculating probabilities for each possible character value at each internal node of the tree; an example is given in figure 4. The probabilities of the various instructions for a given tuple can also be calculated and can be used to estimate instruction probabilities for each machine from a given K-way alignment. All this can be done by traversing the tree, in O(K) time, for each tuple but it is a significant computation and should not be repeated unnecessarily. Therefore results are stored in a lookup-table for fast access if ever the tuple is met again, as it probably will be. The lookup-table speeds up the algorithm considerably. Two alignments of 4 strings of length 500 can be aligned, to give an 8-way alignment, in 15 seconds on a sparc-station.
Sampling Alignments of K > 2 Strings
It is infeasible to average over all K-way alignments of K strings for the purpose of estimating the edges of a given evolutionary tree but it can be sufficient to average over a large sample of alignments. Unfortunately it is also infeasible to extend the alignment sampling procedure of section 3 directly to K>2 strings for the same algorithmic reasons that the DPA cannot be directly extended. However, we can think of an alignment as the state of a complicated system having many degrees of freedom. It is sufficient to hold many of those degrees of freedom fixed while sampling the remainder from the conditional posterior distribution so that is what is done: Given a multiple alignment, the tree is "broken" on a random edge which partitions the strings into two disjoint sets, as in section 4. The multiple alignment is projected onto the two sets of strings to give two subalignments. A random realignment is sampled from the posterior distribution of alignments (of the subalignments) as described for just two strings in section 3 and using the costs for K-tuples as described in section 4. The realignment is sampled conditional on the subalignments. Each subalignment and the degrees of freedom that it specifies remain fixed during the realignment. Only the relation between the two subalignments is sampled in this step but the process is iterated many times, choosing random edges.
The machine parameters are estimated for each multiple alignment sampled. Results from all samples are averaged to give the final estimate of the machine parameters. Standard deviations are also calculated and give an indication of the accuracy of estimates. A working-estimate of the machine parameters is needed to calculate the distributions in the algorithm and a weighted average from "recent" alignments is used for this purpose; the algorithm seems to be insensitive to the details of how this is done. To begin the sampling, an initial multiple alignment is found by the deterministic heuristic described previously.
Simulated Annealing
The alignment sampling procedure of the previous section is trivially modified for use in a simulated annealing approach to optimal multiple alignment. At the heart of the sampling procedure a point j of string or alignment B is chosen to correspond to the midpoint i of string or alignment A. The point j is sampled from the probability distribution of possible values. If the values of the probability distribution are raised to some power p>=0, and the distribution is renormalised, the sampling of j, and hence of alignments, is modified in a useful way. When p=0, j is chosen from a uniform distribution. When p=1, j is chosen from the posterior distribution implied by alignments. When p>1, j is biased towards the more probable values. When p is very large, j is chosen so as to lead to an optimal (pairwise) alignment; the procedure becomes Hirschberg's algorithm in effect. Increasing p implements lowering the temperature in simulated annealing. Since the algorithm actually uses message lengths, the message lengths are multiplied by p which is equivalent to raising the probabilities to the power p.
This strategy is very different in action from the simulated annealing strategy of Ishikawa et al(1992), which makes small perturbations to a multiple alignment. The present strategy can make large perturbations to an alignment, especially when p is small.
Results
A program was written containing the alignment sampling procedure and the simulated annealing method described above. It was first tested on artificially generated DNA data. Figure 5 shows the artificial evolution of three generations of strings. The original ancestor, s1, is a random string of 500 characters. The first-generation descendants are s2 and s3; s3 is identical with s1 to make all edges similar because we are dealing with unrooted trees. Every edge in the tree corresponds to a mutation-machine with the following parameters: P(copy)=0.9, P(change)=0.05; P(insert)= P(delete)=0.025. One expects something like 10% mutation from parent to child but it could be more or less as the evolutionary process is random. One expects something like 20% mutation between s8 and s9 say, 30% mutation between s4 and s6, 40% mutation between s8 and s10 and 50% mutation between s8 and s12. Note that alignments can be found for s8 and s12 with more than 50% matches (their edit-distance is 189 not 250) and that an alignment with 60% to 70% matches can be found even for two random, unrelated DNA sequences of similar length.
In three separate trials, first, second and third-generation strings were used as data - each with the correct evolutionary tree. The tree for trial 3 includes that for trial 2 which includes the trivial tree for trial 1. Three analyses were performed in each trial. First, the deterministic heuristic was used to find a good alignment and parameters were estimated from this alone. The alignment was used as a starting point for the next two analyses. Second, parameters were estimated from 1000 stochastically sampled alignments. Third, simulated annealing was used over 1000 trials with the message length multiplier increasing linearly from 1.0 to 4.0.
The results of these trials are summarised in Table 1. The figures marked actual give information from the evolution of the strings which is unknown to the analysis program. During evolution the machine on each edge had parameters P(copy)=0.9, P(change)=0.05, P(insert)=0.025, P(delete)=0.025 but there is variation and so the actual figures are given.
The figures marked det give the parameter estimates from the putative optimal alignment found by the deterministic heuristic in each trial. There is the beginning of a trend to overestimate P(copy) and, with the exception of e7 and e10, to underestimate P(indel). This is consistent with previous results on two strings (Yee & Allison 1993). In order to avoid repeated qualification in what follows, we often refer to an alignment found by the deterministic heuristic or by simulated annealing as an `optimal alignment' even though it may only be a near optimal alignment.
The figures marked Gibbs give the estimates from the 1000 sampled alignments in each trial. (The standard deviation of the estimate of P(indel) is the largest and is the only one reproduced.) The actual proportion of indels on each edge lies within about two standard deviations of the estimate of P(indel). Note that the standard deviation of the estimates for e1, which is common to each trial, increases as the data gets farther from e1, roughly doubling with each extra generation, and that the estimates for this edge are the worst.
The deterministic heuristic proved hard to beat in the search for an optimal alignment at this moderate level of mutation. It was not beaten by simulated annealing in any of the above trials although simulated annealing found many alignments with a message length just two bits more than that found by the heuristic. From other trials it also seems that 4-way alignment might be rather easy, in that the heuristic was not beaten in several trials. On 8-way alignment with 10% mutation per edge, the heuristic was sometimes beaten, but never by more than a few bits on artificial data. Possibly the simulated annealing was cooled insufficiently or was cooled too quickly. It seems that there is a very large number indeed of near optimal alignments and that the search-space is hardly informative close to them. It would take extremely time consuming tests to map out the alignment landscape thoroughly. The search for the marginally "best" alignment may be rather pointless in any case.
Simulated annealing beat the deterministic heuristic by a significant 34 bits when the level of mutation was increased to 15% per edge. Table 2 gives results for a tree with the topology of figure 5 where the machine on each edge had parameters P(copy)=0.85, P(change)=0.075, P(insert)= P(delete)=0.0375. Strings such as s8 and s12 are only tenuously related here. Estimates are given from single alignments by the deterministic heuristic (det) and simulated annealing (SA) and from stochastic sampling of 1000 alignments (Gibbs). There is an increased tendency for (near) optimal alignments to underestimate P(indel). This effect is most marked on "internal" edges of the tree, as illustrated by the means over different sets of edges. For example, simulated annealing gives an average of 0.025 against a real figure of 0.073 over e2 to e5. Sampling gives an average of 0.070. Note that much of the improvement in message length in going from the heuristic to simulated annealing seems to be due to the latter "explaining away" more indels as changes on the inner edges. (A similar effect has been noted with algorithms under development for the most parsimonious alignment problem.) The standard deviations in sampling's parameter estimates increase with the level of mutation as is to be expected.
Some tests were also done on unbalanced trees with edges of different lengths. Sampling continued to yield better estimates of actual edges although accuracy decreased and standard deviations increased on the longer edges.
Various tests were done to study the asymptotic behaviour of the algorithms and some results are given in table 3. In order to reduce computer time, only the deterministic heuristic was used to find (near) optimal alignments to compare with sampling. Firstly, 10 data sets were generated for the tree of figure 5 at 20% mutation per edge. This is quite a high level of mutation; across the 10 data sets the message lengths range from 7700 to 7900 bits for an optimal alignment, from 8000 to 8150 bits for the null theory and from 9100 to 9900 bits for an average alignment. (Note that the message length of the r-theory, if it could be calculated, would be less than that of an optimal alignment.) Averages over all edges and all 10 data sets of actual and estimated parameters are shown in table 3, line (a). There is an increased tendency for optimal alignments to underestimate P(indel) at the 20% mutation level, particularly on inner edges of the tree. In going from table 2 to table 3 line (a), the average frequency of indels per edge has risen from 0.072 to 0.099 but the estimate from optimal alignment has remained at 0.048. Stochastic sampling gives good average estimates, within 0.01 of the actual figures. However for some data sets the estimates of some edges by sampling have standard deviations of over 0.06 implying that little more than one decimal place of such an estimate may be usable. Subsequently, 10 data sets were generated for the tree of three leaves, (s1 (s2 s3)), at each of 10%, 20% and 30% mutation per edge. Averages over all edges and all 10 data sets of actual and estimated parameters are shown for each level of mutation in table 3 lines (b) to (d). Sampling gives good estimates although standard deviations of estimates rise with the mutation level. At the same level of mutation per edge, the estimates from optimal alignment are better for the three leaf tree than for the 8 leaf tree, presumably because the former has fewer degrees of freedom available for the maximisation of alignment probability. The mutation level of 30% per edge is high and an optimal three-way alignment typically fails to be an acceptable hypothesis by a margin of 30 to 60 bits. The results also suggest that there may be a small bias in the sampling program to overestimate P(indel) at high levels of mutation. This possibility is being further investigated.
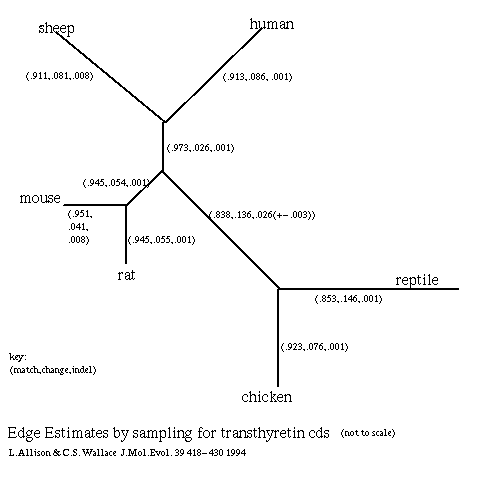
Transthyretin is a protein expressed in the brain. It is also expressed in the liver of some animals. Amongst other things, it is relevant to the evolution of birds, reptiles and mammals (Schreiber et al 1992). The cds sequences for transthyretin (Duan et al 1991) from human, sheep, mouse, rat, chicken and reptile (T.rugosa) were obtained from Genbank. There is little doubt about the correct topology of the evolutionary tree, which is shown in figure 6 annotated with the estimates of the parameters for each edge as averaged from sampling 1000 alignments. There must be considerable pressure of selection on these sequences, some relationships being close, and only the edge joining birds and reptiles to mammals shows significant numbers of indels. The standard deviations of estimates are low as the alignment is constrained. An optimal alignment gives similar estimates.
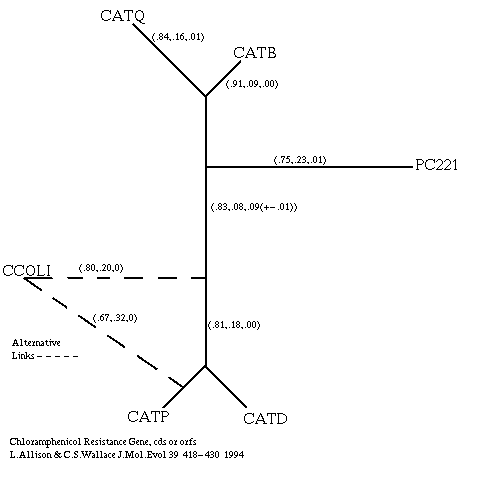
Huggins et al (1992) constructed a tree of chloramphenicol acetyltransferase (CAT) monomers from various bacteria. Tests on five of the corresponding DNA sequences (Allison & Wallace 1994) revealed an interesting possibility. The tree (((CATQ CATB) (CATP CATD)) CCOLI) is weakly preferred on the basis of the message length of an optimal alignment. However, the tree ((CATQ CATB) ((CATP CCOLI) CATD)) is preferred on the basis of the average message length of sampled alignments, although the difference between the trees is less than the sum of the standard deviations in message lengths. This implies some uncertainty in the placing of CCOLI. Subsequently a sixth DNA sequence, PC221, was added to the data set. All test results are clear on how PC221 should be linked to each of the two trees above. For the six strings, the tree (((CATQ CATB) PC221) ((CATP CCOLI) CATD)) is supported over (((CATQ CATB) PC221) ((CATP CATD) CCOLI)) by both the optimal alignment and the average alignment criteria, although only weakly by the former. These trees are also closer, in terms of the average message length of alignments, than the trees on five sequences. As it happens the correct tree is almost certainly (((CATQ CATB) PC221) ((CATP CATD) CCOLI)) which is a subtree of the tree that Huggin's et al inferred using protein sequences and using sequences from more organisms. The situation is illustrated in figure 7. The annotations on the edges come from Gibbs sampling 1000 alignments of the six sequences under either tree as convenient; the two trees had good agreement on common edges. The significance of the above to the particular case of CAT genes is probably not great as the analysis is based on a simple model of evolution that is not the best for coding sequences and takes no account of expert biological knowledge. However, the results do illustrate the important general point that optimal alignment message length and average alignment message length may support different trees. An interesting question is which one should be believed in such cases? The best answer is neither. The best criterion for choosing a tree would be based on the message length of the r-theory: the -log2 probability of getting the strings given the tree, ie. the -log2 of the sum of the probabilities of all alignments given the tree. This could support a different tree entirely. Unfortunately it is infeasible to calculate its message length. The message length of an optimal alignment provides an upper bound on that of the r-theory, and a good alignment contributes more to the probability of the r-theory than an average one contributes, but there are many more of the latter. It is only sensible to exercise caution in such cases.
Conclusions
An alignment of a set of strings can give an estimate of the edges of an evolutionary tree over the strings. However the use of a good or optimal alignment gives a biased estimate of the true values, particularly on the inner edges of the tree. Forming the weighted average of estimates from all alignments would give an unbiased estimate of the edges. This can be done efficiently for two strings but is infeasible for more than two. However, averaging the estimates from many alignments sampled from their posterior probability distribution gives a good approximation and is feasible. In addition, sampling from the probability distribution raised to an increasing power (or from message lengths with an increasing multiplier) effects a simulated annealing search for an optimal alignment. A computer program embodying these ideas has been implemented for the simplest, 1-state model of evolution. We intend to extend the model although this is not trivial for more than two strings. The current implementation is practical for 10 strings of several hundred characters when used on a good work-station. With some optimisation each limit could be increased somewhat. It is tempting to reduce the time-complexity of the underlying DPA from quadratic to near linear by a windowing technique under the assumption that most of the probability in alignment-space is concentrated near the current alignment. However this may be a trap because the assumption may be invalid, particularly if the strings contain repeated and transposed sections. The sampling method is certainly a good candidate for implementation on a parallel computer. The tuning of simulated annealing algorithms is a difficult area and more work needs to be done on tuning the one described here.
It would be useful to be able to handle many more than 10 strings. To do this it will probably be necessary to use a method, related to the one described here, but which samples definite strings for the internal, hypothetical nodes of the tree in a stochastic manner. (The current method makes only implicit, probabilistic assignments to internal nodes.) The problem can then be treated as an iterated three-descendant problem. Each step will be relatively simple but more of them will probably be required to explore the search-space adequately. The resulting program would be a stochastic version of a method proposed by Sankoff et al(1976). A program of this type is under development.
Our sampling and simulated annealing techniques could be used with
other cost functions, such as star and all-pairs costs, in multiple alignment
provided that the costs can be interpreted
as something like the -log2 of a probability.
It would be sufficient for the rank-ordering of the better alignments
to be correct at low temperature.
Simulated annealing could help with the problem of local optima
that also affects alignment under such costs.
However, it is not clear what the results of stochastic sampling would mean
for these costs as they do not seem to have an obvious
evolutionary interpretation.
Appendix: Sampling Alignments of Two Strings
| Also see: L. Allison. Using Hirschberg's algorithm to generate random alignments of strings, Inf. Proc. Lett., 51(5), pp.251-255, Sept. 1994. |
The alignment sampling procedure of section 3
is recursive and is in general called
to act on a substring of string A and a substring of string B.
If either substring is empty there is only one possible alignment.
Otherwise, suppose we are given substrings A[k..m] of string A where m>k
and B[h..n] of string B where n>=h.
Let i be the middle position (k+m)div 2 of A[k..m].
Consider an arbitrary alignment of A[k..m] and B[h..n].
A[i] appears somewhere in the alignment.
It either occurs alone as <A[i],-> which is equivalent to delete(A[i])
or as <A[i],B[j]> for some j which is equivalent to copy(A[i]) or to
change(A[i],B[j]).
In either case let B[j] be the last character of B[h..n], if any,
that occurs with or before A[i]; h-1<=j<=n.
There is a certain probability distribution over the possible values
of j given i.
The probability of a particular value `j' is proportional to the sum
of the probabilities of all alignments that involve its choice
ie. events (i) and (ii) below.
(i) A[k..i-l]&B[h..j]; <A[i], - >; A[i+1..m]&B[j+1..n]
... del A[i] ...
for some j, h-1<=j<=n
(ii) A[k..i-1]&B[h..j-1]; <A[i], B[j]>; A[i+1..m]&B[j+1..n]
... copy A[i] or ...
... change A[i]toB[j] ...
for some j, h<=j<=n
Divide-and-Conquer Cases of A[k..m]&B[l..n], m>k, n>=h.
Now P(A[p..q]&B[r..s]) can be calculated for all `s' by the modified (logplus) DPA of figure 3c. Therefore the probabilities of each possible value of j can be calculated with two calls to the DPA: one on the forward strings A[k..i]&B[h..n] and one on the reversed strings A[i+1..m]&B[h..n]. The forward run is used to calculate the probabilities of A[k..i] and each prefix of B[h..n] being generated together, by any instruction sequence whose final instruction includes A[i]. The reverse run calculates the probabilities of A[i+1..m] and each suffix of B[h..n] being generated together in any way. Combining the results, using logplus, gives the -log2 odds ratios of all possible ways of partitioning an instruction sequence for A and B at the instruction that includes A[i]. This allows j to be sampled from its correct probability distribution. The sampling procedure is then called recursively on A[k..i]&B[h..j] and on A[i+1..m]&B[j+1..n].
The coordinates (i,j) are called an internal terminal. The coordinates (|A|,|B|) are called the external terminal. Since the first half of the overall alignment was forced to end with either <A[i],-> or <A[i],B[j]> and not <-,B[j]>, any subsequent reversed DPAs are forced to begin from an internal terminal with one of these alternatives by modification of the boundary conditions.
The lengths of the sections of A and B shrink with each recursive call to the sampling procedure. Eventually a single character of A remains and this leads to the base cases of the procedure. If the call is for an internal terminal, A[i] must occur at the end of the mini-alignment. There are two possibilities:
(i) - ... - A[i]
B[j] ... B[k] -
ins ... ins del
(ii) - ... - A[i]
B[j] ... B[k-1] B[k]
copy or
ins ... ins change
Internal-Terminal Base-Cases of A[i]&B[j..k], k>=j.
The probability of each possibility is easily calculated and they are sampled accordingly. If the base case is for the external terminal there are more possibilities but each is no more complex than before and they are easily sampled:
(i) A[i] - ... - or - A[i] - ... - etc.
- B[j] ... B[k] B[j] - B[j+1] ... B[k]
del ins ... ins ins del ins ... ins
(ii) A[i] - ... - or - A[i] - ... - etc.
B[j] B[j+1] ... B[k] B[j] B[j+1] B[j+2] ... B[k]
copy/ copy/
chng ins ... ins ins chng ins ... ins
External-Terminal Base-Cases of A[i]&B[j..k].
Essentially the same procedure is used to sample K-way alignments of K-strings as described in section 5.
|
Also see the
[bioinformatics]
pages. |
References
Allison L. & C. S. Wallace. (1994) An information measure for the string to string correction problem with applications. 17th Australian Comp. Sci. Conf., Christchurch, New Zealand, 659-668.
Allison L., C. S. Wallace & C. N. Yee. (1992a)
Finite-state models in the alignment of macro-molecules.
J. Mol. Evol. 35 77-89.
(Also see
[90/148].)
Allison L., C. S. Wallace & C. N. Yee. (1992b) Minimum message length encoding, evolutionary trees and multiple alignment. 25th Hawaii Int. Conf. Sys. Sci. 1 663-674.
Bishop M. J. & E. A. Thompson. (1986) Maximum likelihood alignment of DNA sequences. J. Mol. Biol. 190 159-165.
Duan W., M. G. Achen, S. J. Richardson, M. C. Lawrence, R. E. H. Wettenhall, A. Jaworowski. & G. Schreiber. (1991) Isolation, characterisation, cDNA cloning and gene expression of an avian transthyretin: implications for the evolution of structure and function of transthyretin in vertebrates. Eur. J. Biochem. 200 679-687
Felsenstein J. (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17 368-376.
Felsenstein J. (1983) Inferring evolutionary trees from DNA sequences. In Statistical Analysis of DNA Sequence Data B. S. Weir (ed), Marcel Dekker 133-150.
Hastings W. K. (1970) Monte Carlo sampling methods using Markov chains and their applications. Biometrika 57 97-109.
Haussler D., A. Krogh, S. Mian & K. Sjolander. (1993) Protein modelling using hidden Markov Models: Analysis of globins. 26th Hawaii Int. Conf. Sys. Sci. 1 792-802.
Hirschberg D. S. (1975) A linear space algorithm for computing maximal common subsequences. Comm. A.C.M. 18(6) 341-343.
Huggins A. S., T. L. Bannam & J. I. Rood. (1992) Comparative sequence analysis of the catB gene from clostridium butyricum. Antimicrobial agents and chemotherapy 36(11) 2548-2551.
Ishikawa M., T. Toya, M. Hoshida, K. Nitta, A. Ogiwara & M. Kanehisa. (1992) Multiple sequence alignment by parallel simulated annealing. Institute for New Generation Computing (ICOT) TR-730
Lawrence C. E., S. F. Altschul, M. S. Boguski, J. S. Liu, A. F. Neuwald & J. C. Wooton. (1993) Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science 262 208-214.
Sankoff D., R. J. Cedergren & G. Lapalme. (1976) Frequency of insertion-deletion, transversion, and transition in evolution of 5S ribosomal RNA. J. Mol. Evol. 7 133-149.
Schreiber G., A. R. Aldred & W. Duan. (1992) Choroid plexus, brain protein-homeostasis and evolution. Today's Life Science Sept. 22-28.
Thorne J. L., H. Kishino & J. Felsenstein. (1991) An evolutionary model for maximum likelihood alignment of DNA sequences. J. Mol. Evol. 33 114-124.
Thorne J. L., H. Kishino & J. Felsenstein. (1992) Inching towards reality: an improved likelihood model of sequence evolution. J. Mol. Evol. 34 3-16.
Wallace C. S. & D. M. Boulton. (1968) An information measure for classification. Comp. J. 11(2) 185-194.
Wallace C. S. & P. R. Freeman. (1987) Estimation and inference by compact encoding. J. Royal Stat. Soc. B 49 240-265.
Yee C. N. & L. Allison. (1993) Reconstruction of strings past. Comp. Appl. BioSci. 9(1) 1-7.
string A = TAATACTCGGC string B = TATAACTGCCG mutation instructions: copy change(ch) NB. ch differs from the corr' char in string A. del ins(ch) a mutation sequence: copy; copy; delete; copy; copy; ins(A); copy; copy; del; copy; change(C); copy; ins(G) generation instructions: match(ch) mismatch(chA,chB) NB. chA!=chB insA(chA) insB(chB) a generation sequence: match(T); match(A); insA(A); match(T); match(A); insB(A); match(C); match(T); insA(C); match(G); mismatch(G,C); match(C); insBG equivalent alignment: TAATA-CTCGGC- || || || | | TA-TAACT-GCCG
Figure 1: Basic Models.
P -- hypothetical, unknown parent
. .
mA. .mB -- mutation machines
. .
. .
A B -- given descendants
m = mA.mB -- equivalent generation machine
m's instns Explanation &
pair (a,b) rates per character of P.
------------- --------------------------------------------------
match(x) x<---x--->x x<---y--->x
x x pA'(c).pB'(c) pA'(ch).pB'(ch)/3
mismatch(x,y) x<---x--->y x<---y--->y x<---z--->y
x y pA'(c).pB'(ch) pA'(ch).pB'(c) pA'(ch).pB'(ch)2/3
insA(x) x<---x--->_ x<---y--->_ x<---_--->_
x _ pA'(c).pB'(d) pA'(ch).pB'(d) pA'(i)
insB(x) _<---x--->x _<---y--->x _<---_--->x
_ x pA'(d).pB'(c) pA'(d).pB'(ch) pB'(i)
_<---x--->_ "invisible",
_ _ pA'(d).pB'(d) not seen
c-copy, ch-change, i-insert, d-delete
Define instn rates per character of P:
pA'(instn) = pA(instn)/(1-pA(i)); pB'(instn) = pB(instn)/(1-pB(i))
Note pA'(c)+pA'(ch)+pA'(d) = pB'(c)+pB'(ch)+pB'(d) = 1
Sum rates = (1+pA'(i)+pB'(i)) = (1-pA(i).pB(i))/((1-pA(i)).(1-pB(i)))
Figure 2: Composition of Two Machines, for DNA.
Boundary Conditions, i=1..|A|, j=1..|B|:
D[0,0] = 0
D[i,0] = D[i-1,0]+f(A[i], '-')
D[0,j] = D[0,j-1]+f('-', B[j])
General Step, i=1..|A|, j=1..|B|:
D[i,j] = g( D[i-1,j-1]+f(A[i],B[j]), -- match or mismatch
D[i-1,j ]+f(A[i], '-'), -- insA A[i]
D[i, j-1]+f('-', B[j]) ) -- insB B[j]
(a) For Edit Distance:
f(x,x) = 0; f(x,y) = f(x,'-') = f('-',y) = 1
g(a,b,c) = min(a,b,c)
(b) For most probable alignment:
f(x,x) =-log2(P(match(x))); f(x,y) =-log2(P(mismatch(x,y)));
f(x,'-')=-log2(P(insA(x))); f('-',y)=-log2(P(insB(y)))
g(a,b,c)= min(a,b,c)
(c) For r-theory:
f( , ) as for (b)
g(a,b,c)= logplus(a,logplus(b,c))
where logplus(-log2(p),-log2(q)) = -log2(p+q)
Figure 3: Dynamic Programming Algorithm (DPA) Variations.
Actual machine parameters:
copy change indel
m1: 0.9 0.05 0.05
m2: 0.9 0.08 0.02
m3: 0.7 0.2 0.1
m4: 0.8 0.1 0.1
m5: 0.75 0.1 0.15
A A
. .
m1. .m3
. A:0.43 A:0.40 .
.C:0.56 C:0.59.
+---------------------+
. m5 .
. .
m2. .m4
. .
C C
Probable hypothetical characters.
Tuple ACAC's probability = 0.00027
Estimated operations carried out:
P(copy) P(change) P(indel)
m1: 0.43 0.57 <0.01
m2: 0.57 0.43 <0.01
m3: 0.40 0.60 <0.01
m4: 0.59 0.41 <0.01
m5: 0.94 0.06 <0.01
Figure 4: Example, explanations for tuple ACAC.
s8 s15
. .
s9 . . s14
. . e6 e13 . .
. . . .
e7 . . . . e12
. . . .
. . . .
s4 s7
. .
. .
e2 . . e5
. .
. e1 .
s2..............s1=s3
. .
. .
. e3 . e4
. .
. .
s5 s6
. . . .
. . . .
e8 . . . . e11
. . . .
. . e9 . e10 .
s10 . . s13
. .
s11 s12
Ancestor, s1=s3, length(s1)=500
1st generation: s2 and s3
2nd generation: s4 to s7
3rd generation: s8 to s15
Probabilities for each edge during evolution:
P(copy)=.9; P(change)=.05; P(insert)=P(delete)=.025
Figure 5: Full Tree.
sheep human
. .
. .
(.911,.081,.008) . .(.913,.086,.001)
. .
+
.
.(.973,.026,.001)
+
. .
(.945,.054,.001). .
. .
. .
mouse............+ .(.838,.136,.026(+-.003))
(.951,.041,.008) . .
. .
.(.945,.055,.001) .
. .
. +......................reptile
rat . (.853,.146,.001)
.
.(.923,.076,.001)
.
.
chicken
key: (P(match),P(change),P(indel))
Genbank ids: HUMPALA(27-470), OATTHYRE(12-452), MMALBR(27-467),
RATPALTA(10-453), GDTRTHY(26-478), TRTRANST(16-468).
Figure 6: Edge Estimates by Sampling for Transthyretin cds.
CATQ
.
(.84,.16,.01). CATB
. .
. .(.91,.09,0)
.
.
.(.84,.14,.02)
.
.
.........................PC221
. (.75 .23,.01)
.
.(.83,.08,.09(+-.01))
(.80,.20,0) .
CCOLI--------------.
+ .
+ .(.81,.18,0)
+ .
(.67,.32,0)+ .
+ . .
. . unmarked => P(copy) >= .97
. .
CATP CATD
Message Lengths (bits):
Tree over 5 sequences opt' Gibbs (SD)
----------------------------------- ---- ----------
(((CATQ CATB)(CATP CATD))CCOLI) --- 4369 4555(+-40)
((CATQ CATB)((CATP CCOLI)CATD)) +++ 4376 4502(+-26)
Null: 6449 bits
Tree over 6 sequences opt' Gibbs (SD)
------------------------------------------ ---- ----------
(((CATQ CATB)PC221)((CATP CATD)CCOLI)) --- 5395 5564(+-36)
(((CATQ CATB)PC221)((CATP CCOLI)CATD)) +++ 5391 5519(+-32)
Null: 7752 bits
Data: chloramphenicol resistance gene, cds or orfs from
Genbank-id organism designation
m55620 Clostridium perfringens 459-1118 CATQ
m28717 Clostridium perfringens 145-768 CATP
x15100 Clostridium difficile 91-726 CATD
m35190 Campylobacter coli 309-932 CCOLI
m93113 Clostridium butyricum 145-800 CATB
spc221 Staphylococus aureus 2267-2914 PC221
Figure 7: Alternative Trees for Bacterial Sequences.
(i) data = 1st generation, s2 and s3:
edge actual det Gibbs
---- -------------- -------------- --------------------
e1: .895 .037 .068 .900 .040 .061 .893 .038 .070(.005)
(ii) data = 2nd generation, s4 to s7:
edge actual det Gibbs
---- -------------- -------------- --------------------
e1: .895 .037 .068 .894 .044 .063 .878 .038 .085(.009)
e2: .902 .053 .045 .910 .054 .036 .899 .052 .049(.007)
e3: .919 .041 .039 .916 .057 .026 .917 .048 .035(.005)
e4: .898 .055 .047 .899 .059 .043 .891 .054 .056(.007)
e5: .908 .041 .051 .915 .055 .030 .908 .052 .041(.006)
-------------- -------------- --------------
means over subsets of edges:
e2-5: .907 .048 .046 .910 .056 .034 .904 .052 .045
e1-5: .904 .045 .050 .907 .054 .040 .899 .049 .053
(iii) data = 3rd generation s8 to s15:
edge actual det Gibbs
---- -------------- -------------- --------------------
e1: .895 .037 .068 .878 .060 .062 .863 .043 .094(.016)
e2: .902 .053 .045 .928 .048 .025 .900 .054 .046(.007)
e3: .919 .041 .039 .915 .061 .025 .920 .048 .033(.008)
e4: .898 .055 .047 .920 .038 .043 .906 .036 .058(.009)
e5: .908 .041 .051 .913 .055 .032 .904 .061 .035(.008)
e6: .891 .058 .051 .896 .066 .038 .893 .062 .045(.006)
e7: .926 .035 .039 .924 .035 .042 .918 .031 .052(.007)
e8: .922 .029 .049 .937 .037 .026 .932 .026 .041(.006)
e9: .898 .035 .067 .909 .042 .050 .895 .032 .073(.009)
e10: .893 .060 .048 .898 .048 .055 .879 .052 .069(.009)
e11: .902 .049 .049 .907 .057 .036 .906 .053 .042(.007)
e12: .898 .049 .053 .909 .045 .046 .902 .040 .059(.008)
e13: .916 .049 .035 .912 .064 .024 .906 .064 .030(.007)
-------------- -------------- --------------
means over subsets of edges:
e2-5: .907 .048 .046 .919 .051 .031 .908 .050 .043
e6-13: .906 .046 .049 .912 .048 .040 .904 .045 .051
e1-13: .905 .045 .049 .911 .050 .039 .902 .046 .052
Evolution: Ancestor length = 500
P(copy)=0.9; P(change)=0.05; P(insert)=P(delete)=0.025 for each edge
key: P(copy) P(change) P(indel)
actual = frequencies as measured during evolution
det = inferred from a good alignment by deterministic heuristic
Gibbs = averaged from 1000x sampled alignments
Table 1: Estimated Edges from Evolution at 10% Mutation/Edge.
edge actual det SA Gibbs (S.D.)
---- -------------- -------------- -------------- -------------------
e1 .858 .056 .086 .854 .089 .057 .876 .073 .051 .850 .073 .078(.015)
e2 .849 .073 .079 .862 .093 .046 .869 .080 .051 .838 .067 .095(.017)
e3 .881 .069 .051 .920 .050 .030 .908 .069 .024 .887 .062 .052(.012)
e4 .839 .085 .076 .863 .103 .034 .864 .119 .018 .836 .107 .058(.014)
e5 .833 .079 .088 .845 .133 .022 .833 .160 .008 .836 .089 .075(.019)
e6 .862 .063 .076 .877 .059 .064 .875 .041 .085 .850 .047 .103(.011)
e7 .871 .069 .061 .880 .079 .041 .876 .093 .031 .877 .084 .040(.009)
e8 .885 .049 .066 .890 .062 .049 .896 .053 .051 .890 .053 .057(.010)
e9 .831 .083 .086 .841 .098 .062 .838 .091 .072 .813 .079 .108(.012)
e10 .873 .066 .060 .895 .045 .060 .880 .047 .074 .877 .052 .071(.009)
e11 .837 .088 .074 .851 .090 .059 .860 .074 .065 .836 .075 .089(.013)
e12 .856 .078 .066 .876 .083 .041 .889 .078 .033 .861 .090 .049(.008)
e13 .844 .084 .072 .866 .083 .051 .854 .079 .066 .846 .075 .079(.011)
-------------- -------------- -------------- --------------
means over subsets of edges:
e2-5 :.851 .077 .073 .873 .095 .033 .869 .107 .025 .849 .081 .070
e6-13:.857 .073 .070 .872 .075 .053 .871 .070 .060 .856 .069 .075
e1-13:.855 .072 .072 .871 .082 .047 .871 .081 .048 .854 .073 .073
-------------- -------------- -------------- --------------
Message Lengths (bits):
6735 6701 7556(+-109) mean(SD)
Null: 8045
Evolution: Ancestor length = 500
P(copy)=.85; P(change)=.075; P(insert)=P(delete)=.0375 for each edge
key: frequencies or estimated probabilities of copy, change & indel from
actual - as counted during evolution
det - estimated from a "good" alignment by deterministic heuristic
SA - from an optimal (?) alignment by simulated annealing
Gibbs - from 1000 x Gibbs sampling
Table 2: Estimated Edges from Evolution at 15% Mutation/Edge.
generation actual det Gibbs (std dev's)
---------- -------------- -------------- -----------------------------
a) .8 .1 .1 .801 .101 .099 .828 .125 .048 .790 .107 .103 (.008 to .068)
b) .9 .05 .05 .896 .052 .053 .901 .054 .044 .893 .051 .056 (.003 to .008)
c) .8 .1 .1 .795 .105 .101 .815 .124 .061 .785 .104 .111 (.007 to .022)
d) .7 .15 .15 .702 .150 .148 .745 .194 .061 .683 .141 .176 (.012 to .051)
Ancestor length = 500
key: P(copy) P(change) P(indel)
a) : tree = (((s1 s2)(s3 s4))((s5 s6)(s7 s8))) unrooted
b), c) & d) : tree = (s1 (s2 s3)) unrooted
generation : settings for mutation machine on each edge
actual : as counted during evolution
det : estimated from a "good" or optimal alignment
Gibbs : estimated by Gibbs sampling 1000x per data set
std dev's : range of std dev's for sampling, across 10 data sets
Table 3: Averages over all edges & 10 data sets each for four different settings.
More recent work on Bioinformatics can be found [here]. Inference by [MML] is a general Bayesian method for machine learning and statistical inference. Gibbs sampling is an alternative to Monte Carlo Markov Chain (MCMC) methods when you can sample from the posterior probability distribution.